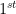JAX
Last year, I had an opportunity to delve into the world of JAX whilst working at InstaDeep. My first blopig post seems like an ideal time to share some of that knowledge. JAX is an experimental Python library created by Google’s DeepMind for applying accelerated differentiation. JAX can be used to differentiate functions written in NumPy or native Python, just-in-time compile and execute functions on GPUs and TPUs with XLA, and mini-batch repetitious functions with vectorization. Collectively, these qualities place JAX as an ideal candidate for accelerated deep learning research [1].
JAX is inspired by the NumPy API, making usage very familiar for any Python user who has already worked with NumPy [2]. However, unlike NumPy, JAX arrays are immutable; once they are assigned in memory they cannot be changed. As such, JAX includes specific syntax for index manipulation. In the code below, we create a JAX array and change the