Two of our esteemed OPIGlets presented a workshop on collaborative research using Jupyter Notebook this week at ISMB in Chicago. Their workshop highlights the importance of finding ways to share your work conveniently and reproducibly. So on a related note, I thought I would share a brief introduction to another useful tool, R Markdown with RStudio, which I use to present updates to various supervisors and to remember what I did three months (or three days) ago. This method of sharing work is highly readable, reproducible, and narrative-driven.
I use R for much of my data analysis and all of my visualisation, and I count the tidyverse among my most beloved friends. If you’re so inclined, it’s easy to execute python, bash, and more from within R Markdown. You also don’t need to use RStudio to use R Markdown, but that’s a whole other story.
Starting a new markdown file in RStudio will generate a template script explaining most of what you need to know. If I showed you that then I’d be out of a blog post, but I will at least link to the R Markdown Reference Guide.
R Markdown files consist of text written in markdown, and code chunks that can be individually executed and displayed inline within RStudio. To “knit” the whole thing together, the knitr package is used to execute and combine code chunks, then pandoc converts the whole thing into an attractive document.
Here’s an example. The metadata at the top sets up the document. I’ll be generating an html document here, but notice some other tempting examples commented out. Yes, you can use it for Latex (swoon). You can even make a Word document, but really, why would you?
---
title: "Informative Title"
author: "Clare E. West"
date: "10/07/2018"
output: html_document
#output: beamer_presentation
#output: pdf_document
---
```{r setup, include=FALSE}
knitr::opts_chunk$set(echo = TRUE)
library(knitr)
library(ggplot2)
library(tidyr)
library(dplyr)
```
## Big Title
### Smaller title
R Markdown scripts have the extension .Rmd
R Markdown is __so__ *fun*. You can read all about it [here](https://www.rstudio.com/wp-content/uploads/2015/03/rmarkdown-reference.pdf).
```{r}
print("Hello world")
```
Notice that chunks are enclosed within three backticks, with the language and options in braces. Single commands can be executed inline using single backticks.
As highlighted in the example above, global options are set like this:
knitr::opts_chunk$set(echo = TRUE)
“echo=TRUE” means that the code in each chunk is displayed in the final product; this is useful to show collaborators (or your future self) exactly how you did something. Change this option (“echo = FALSE”) globally or in individual blocks to prevent code from printing. This is useful to hide uninteresting commands, or when presenting to people who don’t have the time or inclination to read your code (hard to imagine). Notice I’ve also used “include = FALSE” for the library-loading code chunk, which means evaluate but don’t include in the output. Another useful option is “eval = FALSE”, which means don’t even run this chunk.
So let’s see what that looks like when we render it:

The above example output as HTML

The above example output as Latex
Plots generated in code chunks or images from other sources can be embedded. Set the width in the options. “fig.width” sets the width (in inches) of the figure generated, while “out.width” scales the image in the final documents, for which the units will depend on the document type. Within RStudio, these are previewed inline below the code chunk.
## Including plots/images
```{r fig.width = 4, fig.height = 3, out.width = "400px", echo=FALSE}
t %>% group_by(Tour, Winner, N, Tournament) %>% filter(WRank <= 20) %>% summarise(WPts = max(WPts)) %>% ggplot(aes(x=N, y=WPts, group=Winner, colour=(Winner=="Murray A."))) + geom_point() + geom_line() + labs(x="Tournament Number",y="Ranking Points") + scale_colour_discrete("",labels=c("Not Andy Murray", "Andy Murray")) + theme_bw() + theme(legend.position = "bottom", legend.margin = margin(0, 0, 0, 0))
knitr::include_graphics("https://s.yimg.com/ny/api/res/1.2/69ZUzNSMYb09GKd8CNJeew--~A/YXBwaWQ9aGlnaGxhbmRlcjtzbT0xO3c9ODAwO2g9NjAw/http://media.zenfs.com/en_us/News/afp.com/0102e1f7d0d3c35303c8a62d56a5eb79c2c8b4d8.jpg")
```

Rather than just printing data R-style, you can nicely format it into a table using kable (part of knitr). I also style mine using kableExtra, which makes it look nice and gives you extra options. By default tables fill the full width, you can override this using e.g. kable_styling(full.width = FALSE, position = “left”). When making a latex document, use kable(table, booktabs = T, “latex”) to get a (reproducible) latex-style table.

Here’s how to use python and bash. Thanks to the package reticulate, you can even share objects between your R and Python chunks. Exclude reticulate (knitr::opts_chunk$set(python.reticulate=FALSE) if you prefer to keep your languages separate.
### Mix it up with python
```{python}
a='Wow python'
print(a.split()[0])
```
What a wild ride.
### or bash
```{bash, echo=TRUE}
ls | head
```
Oh look, there's our output, ready to share.

Finally, if you hate GUIs – and you know I do – you can ditch the interactive notebook part and just generate documents from R Markdown files like this:
rmarkdown::render("BlogExample.Rmd")

 . However this assumption is often violated in virtual screening. For example, a chemist initially focuses on a series of compounds and the information from this series is used to train a model. For some reasons, the chemist changes their focus on a new, structurally distinct series later on and we would not expect the model to accurately predict the labels in the testing sets. Here, we introduce some methods to address this problem.
. However this assumption is often violated in virtual screening. For example, a chemist initially focuses on a series of compounds and the information from this series is used to train a model. For some reasons, the chemist changes their focus on a new, structurally distinct series later on and we would not expect the model to accurately predict the labels in the testing sets. Here, we introduce some methods to address this problem.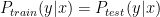 and
and 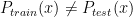 . In general, these methods reweight instances in the training data so that the distribution of training instances is more closely aligned with the distribution of instances in the testing set. The appropriate importance weighting factor
. In general, these methods reweight instances in the training data so that the distribution of training instances is more closely aligned with the distribution of instances in the testing set. The appropriate importance weighting factor 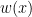 for each instance x in the training set is:
for each instance x in the training set is: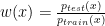
 is the training set density and
is the training set density and 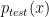 is the testing set density. Note that only the feature vector values (not their labels) are used in reweighting. The major difference between KMM and KLIEP is the objective function: KLIEP is based on the minimisation of the Kullback-Leibler divergence while KMM is based on the minimisation of Maximum Mean Discrepancy (MMD). For more detail, please see reference.
is the testing set density. Note that only the feature vector values (not their labels) are used in reweighting. The major difference between KMM and KLIEP is the objective function: KLIEP is based on the minimisation of the Kullback-Leibler divergence while KMM is based on the minimisation of Maximum Mean Discrepancy (MMD). For more detail, please see reference.




