
While entropy is a major driving force in many chemical changes and is a key component of the free energy of a molecule, it can be challenging to calculate with standard quantum thermochemical methods. With proper consideration in flexible molecules, we can break down the total entropy into different components, including vibrational, translational, rotational and conformational entropy. The calculation of conformational entropy is the most time-consuming as we have to sample all thermally-accessible conformers. Here, we attempt to understand the components that contribute to the conformational entropy of a molecule, and develop a physically-motivated statistical model to rapidly predict the conformational entropies of small molecules.
Continue readingAuthor Archives: Lucian Chan
Understanding the synthesizability of molecules proposed by generative models
De novo molecular design is a computational technique to generate molecules with desired properties from scratch. Classical generative algorithms are based on Genetic Algorithms (GA) and the iterative construction of molecules from molecular fragments. Recently, Variational Auto-Encoders (VAEs), Generative Adversarial Networks (GANs) have been developed for this task, however, the synthesizability of the proposed molecular structures remains an issue. Gao and Coley[1] provided an analysis of the synthesizability of the molecules proposed by these de novo generative algorithms, and discuss their strengths and weaknesses.
Continue readingVisualisation of very large high-dimensional data sets as minimum spanning trees
Large high-dimensional data sets are frequently used in chemical and biological sciences. For example the ChEMBL database contain millions of bioactive molecules from the scientific literature and their associated biological assay data are usually used for drug discovery. Visualising such databases helps understand the structure of data.
Continue readingEffect of Debiasing Protein-Ligand binding data on Generalization
Virtual screening is a computational technique used in drug discovery to search libraries of small molecules in order to identify those structures that bind tightly and specifically to a given protein target. Many machine learning (ML) models have been proposed for virtual screening, however, it is not clear whether these models can truly predict the molecular properties accurately across chemical space or simply overfit the training data. As chemical space contains clusters of molecules around scaffolds, memorising the properties of a few scaffolds can be sufficient to perform well, masking the fact that the model may not generalise beyond close analogue. Different debiasing algorithms have been introduced to address this problem. These algorithms systematically partition the data to reduce bias and provide a more accurate metric of the model performance.
Continue readingBOKEI: Bayesian Optimization Using Knowledge of Correlated Torsions and Expected Improvement for Conformer Generation
In previous blog post, we introduced the idea of Bayesian optimization and its application in finding the lowest energy conformation of given molecule[1]. Here, we extend this approach to incorporate the knowledge of correlated torsion and accelerate the search.
Continue readingFinding the lowest energy conformation of given molecule!
Generating low-energy molecular conformers is important for many areas of computational chemistry, molecular modeling and cheminformatics. Many tools have been developed to generate conformers, including BALLOON (1), Confab (2), FROG2 (3), MOE (4), OMEGA (5) and RDKit (6). The search algorithm implemented in these tools can be broadly classified as either systematic or stochastic. These algorithms primarily focus on generating geometrically diverse low-energy conformers. Here, we are interested in finding lowest energy conformation of a molecule instead of achieving geometric diversity and Bayesian optimization is used to find the lowest energy conformation (7). Continue reading
Covariate Shift in Virtual Screening
In supervised learning, we assume that the training data and testing are drawn from the same distribution, i.e 
Methods such as Kernel Mean Matching (KMM) or Kullback-Leibler Importance Estimation Procedure (KLIEP) have been proposed. These methods typically assume the concept remain unchanged and only the distribution changes, i.e. 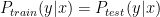
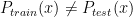
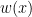
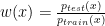
where 
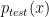
Reference:
- Masashi Sugiyama ,Taiji Suzuki, Shinichi Nakajima, Hisashi Kashima, Paul von Bünau, Motoaki Kawanabe.: Direct importance estimation for Covariate Shift Adaptation. Ann Inst Stat Math. 2008
- Jiayuan Huang, Alex Smola, Arthur Gretton, Karsten Borgwardt, Bernhard Scholkopf.:Correcting Sample Selection Bias by Unlabeled Data. NIPS 06.
-
Mcgaughey, Georgia ; Walters, W Patrick ; Goldman, Brian.: Understanding covariate shift in model performance. F1000Research, 2016,