
The research I do revolves around how to break down protein interaction networks (PINs) into functional modules, or communities, using community detection methods. These communities are groups of proteins which interact more with one another than with the rest of the network. We decompose the PINs in this manner, as it is very difficult to determine how a protein functions through its interactions by simply looking at the entire network. The sheer amount of data in one of these networks, clouds the information we would actually like to see, as explained in my previous blog post.
In a journal club in August, I presented the overlapping community detection method Link Clustering by Ahn et al. which is the first community detection method I am aware of that assigns nodes to communities by clustering the edges between the nodes. I contrasted Link Clustering with the Affiliation Graph Model method proposed by Yang and Leskovec, as the authors use the same comparison technique to validate their methods. In a world where studies usually only work on a single network using a specific method, having two independent papers that use the same validation method is something that gets me much more excited than it probably should.
Link Clustering
Instead of assigning nodes to communities, Link Clustering focusses on the edges in a network. Clustering edges is essentially an equivalent problem to clustering nodes. This becomes obvious if you take a graph, decide to call all the edges “blobs” and say two of your new “blobs” have an edge between them if they share a node in the original network. This is called creating the “line graph” of the network and performing “blob” clustering on this is the same as performing edge clustering on the initial network.
To come back from my tangent, if an edge is assigned to community A, this means the nodes on either side of this edge are assigned community A by association. By assigning each edge to an individual community, Link Clustering creates overlapping node communities as shown in Figure 1 below.
![Example of a simple, link-clustered network. Nodes can be seen to be in multiple communities as edges are assigned to communities (c.f. node 4 in red, green and yellow communities). [Ahn et al., 2010]](https://i0.wp.com/www.blopig.com/blog/wp-content/uploads/2014/10/link_clustering_graph_partition.png?ssl=1)
Figure 1: Example of a simple, link-clustered network. Nodes can be seen to be in multiple communities as edges are assigned to communities (c.f. node 4 in red, green and yellow communities). [Ahn et al., 2010]

Here, 
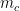


All of this partitioning is based on a similarity score between two edges. The crux of this method, the scoring measure, is explained in Figure 2.
![Schematic Diagram showing how the similarity between edges e_ik and and e_jk is calculated in Link Clustering. The overlap of the inclusive neighbour sets of nodes i and j is divided by the union of these sets. The inclusive neighbour set of node i is computed by taking the neighbours of node i and including node i itself. The nodes in grey are overlapping between both sets, therefore the similarity between edges e_ik and e_jk is 4/12 or 1/3. [Ahn et al 2010]](https://i0.wp.com/www.blopig.com/blog/wp-content/uploads/2014/10/link_clustering_edge_similarity.png?ssl=1)
Figure 2: Schematic Diagram showing how the similarity between edges 
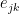
Affiliation Graph Model
The method which I would like to compare Link Clustering to is the Affiliation Graph Model (AGM). Briefly, this is a statistical community detection algorithm which generates overlapping communities. Node affiliations are recorded in the Community Affiliation Matrix shown in Figure 2.
![Schematic of the Affiliation Graph Model, which shows nodes (coloured squares) being affiliated with communities (circles A, B, and C) as shown by the arrows. Nodes can have multiple community affiliations as indicated by red and purple node squares. The probability of interaction between two nodes which are both in only community A, is p_A. [Yang and Leskovec, 2012]](https://i0.wp.com/www.blopig.com/blog/wp-content/uploads/2014/10/AGM_model.png?ssl=1)
Schematic of the Community Affiliation Matrix, which shows nodes (coloured squares) being affiliated with communities (circles A, B, and C) as shown by the arrows. Nodes can have multiple community affiliations as indicated by red and purple node squares. The probability of interaction between two nodes which are both in only community A, is 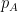
for community 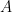 . For nodes
. For nodes  and
and  , which may share several community affiliations, the probability of interaction,
, which may share several community affiliations, the probability of interaction,  is given by the equation:
is given by the equation:
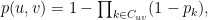
where 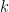

Result Comparison Method
Link Clustering itself has some very interesting results, especially relating to looking at real-world networks at different resolutions (which can be found in the paper). However, I want to focus on the validation method used in the paper which compares results generated from Link Clustering with that of other methods. This comparison method evaluates four different aspects of the partitions generated by different methods. These four aspects are: Community Quality, Overlap Quality, Community Coverage, and Overlap Coverage. The “Quality” aspects relate to network metadata indicating how good the obtained communities are, and the “Coverage” aspects relate to the amount of information extracted from a network.
The metadata referred to above is a general term for additional data available about the nodes in a network. In the case of PINs this metadata is related to the function of proteins in the network (their Gene Ontology annotations). Loosely, proteins with similar functions should be placed in the same community to improve the Community Quality measure. Overlap Quality concerns the boundaries between generated communities. If the functions assigned to proteins show similar boundaries between groups of functions, the Overlap Quality is high.
The “Coverage” values are more basic calculations. A partition has a high Community Coverage if the fraction of nodes assigned to communities with three or more nodes is high (non-trivial communities). A community of size two is uninformative, as it is the result of a single edge being in a community by itself. Overlap Coverage is simply the number of non-trivial community memberships per node. The two “Coverage” values are thus equal for non-overlapping community detection methods.
When comparing community detection methods, these four measures are computed for a partition generated by each method, and then rescaled so that the maximum score achieved by any method is 1 for each measure independently. These values are then added to give a score between 0 and 4 for each community detection method.
Results
The results shown in Figure 4 were generated by Yang and Leskovec to show their AGM outperforms Link Clustering (and other methods) on a variety of networks. While it is noteable that they used the same metric and networks used by Ahn et al. when proposing Link Clustering, this Figure must be taken with a pinch of salt. Yang and Leskovec acknowledge that the metric proposed by Ahn et al. rewards methods which find more communities in a network and thus do not fit the number of communities judged to be best by the AGM, but instead fit the same number as fitted in Link Clustering. Furthermore, it is peculiar that half of the networks used for comparison by Ahn et al. have disappeared in this comparison.
![Community detection method comparison for Link Clustering (L), Clique Percolation (C), Mixed-Membership Stochastic Block Model (M) and the Affiliation Graph Model (A) on different PINs (PPI) and other social networks. Methods are compared by the metric proposed by Ahn et al. [Yang & Leskovec 2012]](https://i0.wp.com/www.blopig.com/blog/wp-content/uploads/2014/10/Model_network_comparison_results.png?ssl=1)
Figure 4: Community detection method comparison for Link Clustering (L), Clique Percolation (C), Mixed-Membership Stochastic Block Model (M) and the Affiliation Graph Model (A) on different PINs (PPI) and other social networks. Methods are compared by the metric proposed by Ahn et al. [Yang & Leskovec 2012]
