
The group asked me if I would tell them a little bit about one of my other hats at our regular Tuesday meeting, and this blog is about that.
In October 2019 I was seconded part-time to UKRI as the Deputy Executive Chair of the Engineering and Physical Sciences Research council (EPSRC). What is UKRI (UK research and Innovation)? It’s a non-departmental public body that funds research and innovation. It is made up of the seven disciplinary research councils (acronyms to please Tom – AHRC, BBSRC, EPSRC, ESRC, NERC, STFC and MRC), Research England, and the UK’s innovation agency, Innovate UK.
As Deputy Executive Chair of EPSRC I was helping with UKRI strategy, learning how a spending review round works, visiting universities to talk about how they could work better with UKRI – pretty much everything I was expecting to be doing. But like everyone, my world changed in early 2020.
Continue reading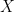 into two sets – a training set
into two sets – a training set 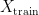 and a test set
and a test set 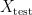 . The model is then subsequently trained on the examples in the training set
. The model is then subsequently trained on the examples in the training set 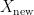 which it will encounter in the future. Right?
which it will encounter in the future. Right?