
Today’s post builds on my earlier blogpost on how to turn a SMILES string into an extended-connectivity fingerprint using RDKit and describes an interesting and easily implementable modification of the extended-connectivity fingerprint (ECFP) featurisation. This modification is based on representing the atoms in the input compound at a different (and potentially more useful) level of abstraction.
We remember that each binary component of an ECFP indicates the presence or absence of a particular circular subgraph in the input compound. Circular subgraphs that are structurally isomorphic are further distinguished according to their inherited atom- and bond features, i.e. two structurally isomorphic circular subgraphs with distinct atom- or bond features correspond to different components of the ECFP. For chemical bonds, this distinction is made on the basis of simple bond types (single, double, triple, or aromatic). To distinguish atoms, standard ECFPs use seven features based on the Daylight atomic invariants [1]; but there is also another less commonly used and often overlooked version of the ECFP that uses pharmacophoric atom features instead [2]. Pharmacophoric atom features attempt to describe atomic properties that are critical for biological activity or binding to a target protein. These features try to capture the potential for important chemical interactions such as hydrogen bonding or ionic bonding. ECFPs that use pharmacophoric atom features instead of standard atom features are called functional-connectivity fingerprints (FCFPs). The exact sets of standard- vs. pharmacophoric atom features for ECFPs vs. FCFPs are listed in the table below.

In RDKit, ECFPs can be changed to FCFPs extremely easily by changing a single input argument. Below you can find a Python/RDKit implementation of a function that turns a SMILES string into an FCFP if use_features = True and into an ECFP if use_features = False.
# import packages
import numpy as np
from rdkit.Chem import AllChem
# define function that transforms a SMILES string into an FCFP if use_features = True and into an ECFP if use_features = False
def FCFP_from_smiles(smiles,
R = 2,
L = 2**10,
use_features = True,
use_chirality = False):
"""
Inputs:
- smiles ... SMILES string of input compound
- R ... maximum radius of circular substructures
- L ... fingerprint-length
- use_features ... if true then use pharmacophoric atom features, if false then use standard DAYLIGHT atom features
- use_chirality ... if true then append tetrahedral chirality flags to atom features
Outputs:
- np.array(feature_list) ... FCFP/ECFP with length L and maximum radius R
"""
molecule = AllChem.MolFromSmiles(smiles)
feature_list = AllChem.GetMorganFingerprintAsBitVect(molecule,
radius = R,
nBits = L,
useFeatures = use_features,
useChirality = use_chirality)
return np.array(feature_list)
The use of pharmacophoric atom features makes FCFPs more specific to molecular interactions that drive biological activity. In certain molecular machine-learning applications, replacing ECFPs with FCFPs can therefore lead to increased performance and decreased learning time, as important high-level atomic properties are presented to the learning algorithm from the start and do not need to be inferred statistically. However, the standard atom features used in ECFPs contain more detailed low-level information that could potentially still be relevant for the prediction task at hand and thus be utilised by the learning algorithm. It is often unclear from the outset whether FCFPs will provide a substantial advantage over ECFPs in a given application; however, given how easy it is to switch between the two, it is almost always worth trying out both options.
[1] Weininger, David, Arthur Weininger, and Joseph L. Weininger. “SMILES. 2. Algorithm for generation of unique SMILES notation.” Journal of Chemical Information and Computer Sciences 29.2 (1989): 97-101.
[2] Rogers, David, and Mathew Hahn. “Extended-connectivity fingerprints.” Journal of Chemical Information and Modeling 50.5 (2010): 742-754.


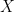 into two sets – a training set
into two sets – a training set 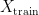 and a test set
and a test set 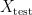 . The model is then subsequently trained on the examples in the training set
. The model is then subsequently trained on the examples in the training set 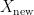 which it will encounter in the future. Right?
which it will encounter in the future. Right?