
Understanding what’s going on when you’ve started training your shiny new ML model is hard enough. Will it work? Have I got the right parameters? Is it the data? Probably. Any tool that can help with that process is a Godsend. Weights and biases is a great tool to help you visualise and track your model throughout your production cycle. In this blog post, I’m going to detail some basics on how you can initialise and use it to visualise your next project.
Installation
To use weights and biases (wandb), you need to make an account. For individuals it is free, however, for team-oriented features, you will have to pay. Wandb can then be installed using pip or conda.
$ conda install -c conda-forge wandb or $ pip install wandb
To initialise your project, import the package, sign in, and then use the following command using your chosen project name and username (if you want):
import wandb wandb.login() wandb.init(project='project1')
In addition to your project, you can also initialise a config dictionary with starting parameter values:
Continue reading

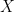 into two sets – a training set
into two sets – a training set 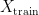 and a test set
and a test set 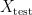 . The model is then subsequently trained on the examples in the training set
. The model is then subsequently trained on the examples in the training set 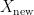 which it will encounter in the future. Right?
which it will encounter in the future. Right?