
This week’s paper by Willis et al. sought to investigate how our limited antibody-encoding gene repertoire has the ability to recognise the unlimited array of antigens. There is a finite number of V, D, and J genes that encode our antibodies, but it still has the capacity to recognise an infinite number of antigens. Simply, the authors’ notion is that an antibody from the germline (via V(D)J recombination; see entry by James) is able to adopt multiple conformations, thus allowing the antibody to bind multiple antigens.

Three antibodies derived from the germline gene 5*51-01 bind to very different antigens.
To test this hypothesis, the authors performed a multiple sequence alignment for the amino acid sequence between the mature antibodies and the germline antibody sequence from which the antibodies are derived from. if a single position from ONE mature antibody showed a difference to the germline sequence, it was identified as a ‘variable’ position, and allowed to be changed by Rosetta’s multi-state design (MSD) and single-state design (SSD) protocols.

Figure 1) from Willis et al., showing the pipeline: align mature antibodies (2XWT, 2B1A, 3HMX) to the germline sequence (5-51) , identify ‘variable’ positions from the alignment, then allow Rosetta to change those residues.
Surprisingly, without any prior information of the germline sequence, the MSD yielded a sequence that was closer to the germline sequence, and the SSD for each mature antibody had retained the mature sequence. In short, this indicated that the germline sequence is a harmonising sequence that can accommodate the conformations of each of the mature antibodies (as proven by MSD), whereas the mature sequence was the lowest energy amino acid sequence for the particular antibody’s conformation (as proven by SSD).
To further demonstrate that the germline sequence is indeed the more ‘flexible’ sequence, the authors then aligned the mature antibodies and determined the deviation in ψ-ϕ angles at each of the variable positions that were used in the Rosetta study. They found that the ψ-ϕ angle deviation in the positions that recovered to the germline residue was much larger than the other variable positions along the antibody. In other words, for the positions that tend to return to the germline amino acid in MSD, the ψ-ϕ angles have a much larger degree of variation compared to the other variable positions, suggesting that the positions that returned to the germline amino acid are prone to lots of movement.
In addition to the many results that corroborate the findings mentioned in this entry, it’s neat that the authors took a ‘backwards’ spin to conventional antibody design. Most antibody design regimes aim to find amino acid(s) that give the antibody more ‘rigidity’, and hence, mature its affinity, but this paper went against the norm to find the most FLEXIBLE antibody (the most likely germline predecessor*). Effectively, they argue that this type of protocol can be exported to extract new antibodies that can bind to multiple antigens, thus increasing the versatility of antibodies as potential therapeutic agents.



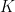 communities and that each node
communities and that each node 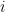 is associated with a vector of community memberships
is associated with a vector of community memberships 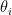 . To generate a network, the model considers each pair of nodes. For each pair
. To generate a network, the model considers each pair of nodes. For each pair 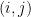 , it chooses a community indicator
, it chooses a community indicator 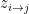 from the
from the 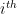 node’s community memberships
node’s community memberships 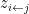 from
from  . A connection is then drawn between the nodes with probability
. A connection is then drawn between the nodes with probability 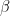 if the indicators point to the same community or with a smaller probability
if the indicators point to the same community or with a smaller probability  if they do not.
if they do not.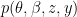 where
where 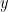 is the observed data. To estimate the posterior
is the observed data. To estimate the posterior 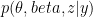 , the method uses a stochastic variational inference algorithm. This enables posterior estimation using only a sample of all-possible node pairs at each step of the variational inference, making the method applicable to very large graphs (e.g analyzing a large citation network of physics papers shown in the figure below identifies important papers impacting several sub-disciplines).
, the method uses a stochastic variational inference algorithm. This enables posterior estimation using only a sample of all-possible node pairs at each step of the variational inference, making the method applicable to very large graphs (e.g analyzing a large citation network of physics papers shown in the figure below identifies important papers impacting several sub-disciplines).
 ) that maximises the following posterior distribution for the number of bins:
) that maximises the following posterior distribution for the number of bins:
 is the number of bins,
is the number of bins,  is the data,
is the data, 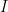 is prior knowledge about the problem, i.e. in particular the use of equal length bins and the range of data
is prior knowledge about the problem, i.e. in particular the use of equal length bins and the range of data 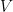 , which has the relation
, which has the relation  where
where  is the width of bins,
is the width of bins, 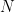 is the number of data points and
is the number of data points and 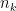 is the number of observations that fall in the
is the number of observations that fall in the 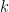 th
th  ) of the bins of the histogram is given by:
) of the bins of the histogram is given by: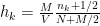 .
.




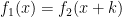 where k is the shift between the two distributions, thus if k=0 then the two populations are actually the same one. This test is based in the rank of the observations of the two samples, which means that it won’t take into account how big the differences between the values of the two samples are, e.g. if performing a WMW test comparing S1=(1,2) and S2=(100,300) it wouldn’t differ of comparing S1=(1,2) and S2=(4,5). Therefore when having a small sample size this is a great loss of information.
where k is the shift between the two distributions, thus if k=0 then the two populations are actually the same one. This test is based in the rank of the observations of the two samples, which means that it won’t take into account how big the differences between the values of the two samples are, e.g. if performing a WMW test comparing S1=(1,2) and S2=(100,300) it wouldn’t differ of comparing S1=(1,2) and S2=(4,5). Therefore when having a small sample size this is a great loss of information.