Predicting the possible shapes a small molecule can adopt is essential to understanding its chemistry and the possible biological roles of candidate drugs. Thus, conformer generation, the process of converting a topological description of a molecule into a set of 3D positions of its constituent atoms, is an essential component of computational drug discovery.
A former member of OPIG, Dr Jean-Paul Ebejer, myself, and Prof. Charlotte Deane, compared the ability of freely-available conformer generation methods:
“(i) to identify which tools most accurately reproduce experimentally determined structures;
(ii) to examine the diversity of the generated conformational set; and
(iii) to benchmark the computational time expended.”
in Ebejer et al., 2012. JP assembled a set of 708 crystal structures of drug-like molecules from the OMEGA validation set and the Astex Diverse Set with which to test the various methods. We found that RDKit, combining its Distance Geometry (DG) algorithm with energy minimization using the MMFF94 force field proved to be a “valid free alternative to commercial, proprietary software”, and was able to generate “a diverse and representative set of conformers which also contains a close conformer to the known structure”.
Following on from our work at InhibOx, and building on the same benchmark set JP assembled, Greg Landrum and Sereina Riniker recently described (Riniker & Landrum, 2015) two new conformer generation methods, ETDG and ETKDG, that improve upon the classical distance geometry (DG) algorithm. They do this by combining DG with knowledge of preferred torsional angles derived from experimentally determined crystal structures (ETDG), and also by further adding constraints from chemical knowledge, such as ‘aromatic rings are be flat’, or ‘bonds connected to triple bonds are colinear’ (ETKDG). They compared DG, ETDG, ETKDG, and a knowledge-based method, CONFECT, and found:
“ETKDG was found to outperform standard DG and the knowledge-based conformer generator CONFECT in reproducing crystal conformations from both small-molecule crystals (CSD data set) and protein−ligand complexes (PDB data set). With ETKDG, 84% of a set of 1290 small-molecule crystal structures from the CSD could be reproduced within an RMSD of 1.0 Å and 38% within an RMSD of 0.5 Å. The experimental torsional-angle preferences or the K terms alone each performed better than standard DG but were not sufficient to obtain the full performance of ETKDG.
Comparison of ETKDG with the DG conformers optimized using either the Universal Force Field (UFF) or the Merck Molecular Force Field (MMFF) showed different results for the two data sets. While FF-optimized DG performed better on the CSD data set, the two approaches were comparable for the PDB data set.”
They also showed (Fig. 13) that their ETKDG method was faster than DG followed by energy minimization, but not quite as accurate in reproducing the crystal structure.
ETKDG takes 3 times as long as DG. The addition of the K terms, i.e., generating ETKDG embeddings instead of ETDG embeddings, increases runtime by only 10% over ETDG (results not shown). Despite the longer runtime per conformer, ETKDG requires on average one-quarter of the number of conformers to achieve performance similar to DG (Figure S12 and Table S4 in the Supporting Information). This results in a net performance improvement, at least when it comes to reproducing crystal conformers.
As measured by performance in reproducing experimental crystal structures, ETKDG is a viable alternative to plain DG followed by a UFF-optimization, so it is of interest how their runtimes compare. Figure 13 (right) plots the runtime for ETKDG versus the runtime for DG + UFF-optimization. The median ratio of the DG + UFF optimization and ETKDG runtimes is 1.97, i.e., DG + UFF optimization takes almost twice as long as ETKDG. Thus, although ETKDG is significantly slower than DG on a per-conformer basis, when higher-quality conformations are required it can provide structures that are the equivalent of those obtained using DG + UFF-optimization in about half the time.
ETKDG looks like a great addition to the RDKit toolbox for conformer generation (and it was great to see JP thanked in the Acknowledgments!).
References
Ebejer, J. P., G. M. Morris and C. M. Deane (2012). “Freely available conformer generation methods: how good are they?” J Chem Inf Model, 52(5): 1146-1158. 10.1021/ci2004658.
Riniker, S. and G. A. Landrum (2015). “Better Informed Distance Geometry: Using What We Know To Improve Conformation Generation.” J Chem Inf Model, 55(12): 2562-2574. 10.1021/acs.jcim.5b00654.

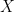 into two sets – a training set
into two sets – a training set 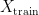 and a test set
and a test set 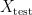 . The model is then subsequently trained on the examples in the training set
. The model is then subsequently trained on the examples in the training set 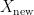 which it will encounter in the future. Right?
which it will encounter in the future. Right?