
… a lot of useful presents as can be seen above … and yes our Christmas party is always in Januray.

… a lot of useful presents as can be seen above … and yes our Christmas party is always in Januray.
Last week I presented a paper by Klei et al. on a new module in the Phenix software suite. This module, entitled Guided Ligand-Replacement (GLR), aims to make it easier to place ligands during the crystallographic model-building process by using homologous models of the ligand-protein complex for the initial placement of the ligand.
In the situation where ligands are being added to a crystallographic protein model, a crystallographer must first build the protein model, identify the difference electron density, and then build the ligand into this density.
The GLR approach is particularly helpful in several cases:
Program Description:
The required inputs for GLR are standard, as required by any ligand-fitting algorithm, namely:
Overview of the program:

Fig 1. Program Overview.
> Identification of the reference structure
Firstly, the program must determine the reference structure to be used as a template. This can be specified by the user, or GLR can search a variety of sources to find the best template. The template selection process is outlined below. Reference structures are filtered by the protein sequence identity, similarity of the molecular weights of the ligands, and finally by the similarity of the binary chemical fingerprints of the ligands (as calculated by the Tanimoto coefficient).

Fig 2. Reference Structure selection flow diagram.
Little justification is given for these cutoffs, although it is generally accepted that proteins with above 70% sequence identity are highly structurally similar. The Tanimoto coefficient cutoff of 0.7 presumably only serves to remove the possibly of very low scoring matches, as if multiple potential reference structures are available, the highest Tanimoto-scored ligand-match is used. They do not, however, say how they balance the choice in the final stage where they take the ligand with the highest Tanimoto score and resolution.
The method for assigning the binary chemical fingerprints can be found here (small error in link in paper).
> Superposition of Reference and Target structures
Once a reference structure has been selected, GLR uses graph-matching techniques from eLBOW to find the correspondences between atoms in the reference and target ligands. These atomic mappings are used to orient and map the target ligand onto the reference ligand.
Once the reference protein-ligand structure is superposed onto the target protein, these atomic mappings are used to place the target ligand.
The target complex then undergoes a real-space refinement to adjust the newly-placed ligand to the electron density. This allows the parts of the target ligand that differ from the reference ligand to adopt the correct orientation (as they will have been orientated arbitrarily by the graph-matching and superposition algorithms).
> Summary, Problems & Limitations
GLR allows the rapid placement of ligands when a homologous complex is available. This reduces the need for computationally intensive ligand-fitting programs, or for tedious manual building.
For complexes where a homologous complex is available, GLR will be able to quickly provide the crystallographer with a potential placement of the ligand. However, at the moment, GLR does not perform any checks on the validity of the placement. There is no culling of the placed ligands based on their agreement with the electron density, and the decision as to whether to accept the placement is left to the crystallographer.
As the authors recognise in the paper, there is the problem that GLR currently removes any overlapping ligands that are placed by the program. This means that GLR is unable to generate multiple conformations of the target ligand, as all but one will be removed (that which agrees best with the electron density). As such, the crystallographer will still need to check whether the proposed orientation of the ligand is the only conformation present, or whether they must build additional models of the ligand.
As it is, GLR seems to be a useful time-saving tool for crystallographic structure solution. Although it is possible to incorporate the tool into automated pipelines, I feel that it will be mainly used in manual model-building, due to the problems above that require regular checking by the crystallographer.
There are several additions that could be made to overcome the current limits of the program, as identified in the paper. These mainly centre around generating multiple conformations and validating the placed ligands. If implemented, GLR will become a highly useful module for the solution of protein-ligand complexes, especially as the number of structures with ligands in the PDB continues to grow.
This week’s paper by Willis et al. sought to investigate how our limited antibody-encoding gene repertoire has the ability to recognise the unlimited array of antigens. There is a finite number of V, D, and J genes that encode our antibodies, but it still has the capacity to recognise an infinite number of antigens. Simply, the authors’ notion is that an antibody from the germline (via V(D)J recombination; see entry by James) is able to adopt multiple conformations, thus allowing the antibody to bind multiple antigens.

Three antibodies derived from the germline gene 5*51-01 bind to very different antigens.
To test this hypothesis, the authors performed a multiple sequence alignment for the amino acid sequence between the mature antibodies and the germline antibody sequence from which the antibodies are derived from. if a single position from ONE mature antibody showed a difference to the germline sequence, it was identified as a ‘variable’ position, and allowed to be changed by Rosetta’s multi-state design (MSD) and single-state design (SSD) protocols.

Figure 1) from Willis et al., showing the pipeline: align mature antibodies (2XWT, 2B1A, 3HMX) to the germline sequence (5-51) , identify ‘variable’ positions from the alignment, then allow Rosetta to change those residues.
Surprisingly, without any prior information of the germline sequence, the MSD yielded a sequence that was closer to the germline sequence, and the SSD for each mature antibody had retained the mature sequence. In short, this indicated that the germline sequence is a harmonising sequence that can accommodate the conformations of each of the mature antibodies (as proven by MSD), whereas the mature sequence was the lowest energy amino acid sequence for the particular antibody’s conformation (as proven by SSD).
To further demonstrate that the germline sequence is indeed the more ‘flexible’ sequence, the authors then aligned the mature antibodies and determined the deviation in ψ-ϕ angles at each of the variable positions that were used in the Rosetta study. They found that the ψ-ϕ angle deviation in the positions that recovered to the germline residue was much larger than the other variable positions along the antibody. In other words, for the positions that tend to return to the germline amino acid in MSD, the ψ-ϕ angles have a much larger degree of variation compared to the other variable positions, suggesting that the positions that returned to the germline amino acid are prone to lots of movement.
In addition to the many results that corroborate the findings mentioned in this entry, it’s neat that the authors took a ‘backwards’ spin to conventional antibody design. Most antibody design regimes aim to find amino acid(s) that give the antibody more ‘rigidity’, and hence, mature its affinity, but this paper went against the norm to find the most FLEXIBLE antibody (the most likely germline predecessor*). Effectively, they argue that this type of protocol can be exported to extract new antibodies that can bind to multiple antigens, thus increasing the versatility of antibodies as potential therapeutic agents.
This week I presented a paper by Burkovitz et al from Bar Ilan University in Israel. The study investigates the mutations that occur in B-cell maturation and how the propensity for a change to be selected is affected by where in the antibody structure it is located. It nicely combines analysis of both DNA and amino-acid sequence with structural considerations to inform conclusions about how in vivo affinity maturation occurs.
Before being exposed to an antigen, an antibody has a sequence determined by a combination of genes (V and J for the light chain; V, D and J for the heavy chain). Once exposed, B-cells (the cells that produce antibodies), undergo somatic hyper-mutation (SHM) to optimise the antibody-antigen (ab-ag) interaction. These mutations are commonly thought to be promoted at activation-induced deaminase (AID) hotspots.
The authors’ first finding is that the locations of SHMs do not correlate well with the positions of AID hotspots and that the distribution of their distance to a hotspot is not much different to that of the background distribution. They conclude that although perhaps a mechanism to promote mutation, AID hotspots are not a strong factor that indicate whether a mutation will fix.
Motivated to find other determinants for SHM preferences, the study turns to examining structural features and energetics of the molecules. SHMs are found to be more prevalent on the VH domain of an Fv than the VL. However, when present, the energetic importance of an SHM is not related to the domain it is on. In contrast, the contribution an SHM makes to the binding energy is related to its structural location. As one might perhaps expect, those SHMs in positions that can make contact with the antigen have more affect than those that do not. Consideration of their propensity instead of raw frequency also shows that SHMs are more prevalent in antibody-antigen interfaces than in the rest of the molecule. However, they are also likely to occur in the VH-VL interface suggesting an importance for this region in fine-tuning the geometry and flexibility of the binding site.

Figure taken from Burkovitz et al shows a) the location of different structural regions on the Fv b) the energetic contribution of the SHMs in each region c) the fraction of SHMs in the regions and their relative size d) the propensity for an SHM to occur in each of the five structural regions.
Perhaps the most interesting result of this study is the authors’ conclusions about the propensity of SHMs to mutate germline residues to particular amino-acids. It is found that whilst germline amino-acid usage in binding sites is distinctive from other protein-protein interfaces, the residue profiles of SHMs are less diverged. They therefore act to bring the properties ab-ag interaction towards those seen in normal interactions. This may suggest, as proposed by other studies, that the somatic hyper-mutation process is similar to mutation properties observed in evolution. In addition, it is found that five amino-acids, asparagine, arginine, serine, threonine and aspartic acid are the most common substitutions made in SHM. Finally, positions where SHMs most often have an important effect on binding energy are presented. These positions, and the amino-acid preferences provide promising targets for use in rational antibody design procedures.
For journal club this week I decided to look at a paper by Moore et al. on the modular evolution of proteins.
Modular evolution, or the rearrangement of the domain architecture of a protein, is one of the key drivers behind functional diversification in the protein universe. I used the example in my talk of the multi-domain protein Peptidase T, which contains a catalytic domain homologous to Carboxypeptidase A, a zinc dependent protease. The additional domain in Peptidase T induces the formation of a dimer, which restricts the space around the active site and so affects the specificity of the enzyme.

The multi-domain protein Peptidase T in a dimer (taken from Bashton and Chothia 2007). The active site is circled in green. Carboxypeptidase A is made up of a single domain homologous to the catalytic domain (in blue) of Peptidase T.
I took this case study from a really interesting paper, The generation of new protein functions by the combination of domains (Bashton and Chothia, 2007), which explores several other comparisons between the functions of multi-domain proteins and their single domain homologues.
What this paper does not address however is the directionality of such domain reorganisations. In all these examples, it is not clear whether the multi-domain organisation has evolved from the single domain enzyme or vice versa. Which brings me back to the paper I was presenting on, which attempts a reconstruction of domain arrangements followed by a categorisation of rearrangement events.
Essentially, given a phylogenetic tree of 20 closely related pancrustacean species, the paper takes the different domain arrangements on the genomes (step 1), assigns the presence or absence of each arrangement at interior nodes on the tree (step 2), and then assigns each gained arrangement to one of four possible rearrangement events (step 3).
1. Domain Annotation
The authors use different methods to annotate domains on the genomes. They conclude the most effective methodology is to use the clan level (where families with suspected homologies are joined together… similar to our beloved superfamily classification) of Pfam-A (high quality family alignments with a manually chosen seed sequence). Moreover, they collapse any consecutive stretches of identical domains into one “pseudo-domain”, eliminating the effect of the domain repeat number on an arrangement’s definition.
2. Ancestral State Reconstruction
The ancestral state reconstruction of each domain arrangement (it’s presence/absence at each internal node on the tree) is a result of a 2-pass sweep across the tree: the first from leaves to root, and the second from the root to the leaves. On the first pass the presence of an arrangement on a parent node is decided by majority rule on the state of its children. If the arrangement is present in one child node but absent in the other, the state at the parent node is defined as uncertain. Any uncertain child nodes have a neutral impact on their parent node’s state (i.e. if a parent has a child with the arrangement and a child with an uncertain state the arrangement will be annotated as present in the parent node). On the second pass (from root to leaves) uncertain nodes are decided by the state at their parent node. An uncertain arrangement at the root will be annotated as present. For more details and a clearer explanation see Box 1 in the figure below.

A schematic for the assignment of domain recombination events. Box 1 gives the algorithm for the ancestral state reconstruction. Figure S2 from Moore et al. 2013.
3. Rearrangement events
For each gained event on a particular branch, the authors then postulated one of four simple rearrangement events dependent on the arrangements on the parent’s predicted proteome.
i) Fusion: A gained domain arrangement (A,B,C) on a child’s proteome is the result of fusion if the parent’s proteome contains both the arrangements (A,B) AND (C) (as one example).
ii) Fission: A gained arrangement (A,B,C) is the result of fission if the parent contains the arrangement (A,B,C,D) AND the child also contains the arrangement (D).
iii) Terminal Loss: A gained arrangement (A,B,C) is the result of terminal loss if the parent contains the arrangement (A,B,C,D) AND the child does not contain the arrangement (D).
iv) Domain gain: A gained arrangement (A,B,C) is the result of domain gain if the parent contains (A,B) but not (C).
Any gained arrangement which cannot be explained by these cases (as a single-step solution) is annotated as having no solution.
Results
The authors find, roughly speaking, that the domain arrangements they identify fall into a bimodal distribution. The vast majority are those which are seen on only one genome, of which over 80% are multi-domain arrangements. There are also a sizeable number of arrangements seen on every single genome, the vast majority of which are made up of a single domain. I do wonder though, how much of this signal is due to the relative difficulty of identifying and assigning multiple different domains compared to just a single domain. While it seems unlikely that this would explain the entirety of this observation (on average, 75% of proteins per genome were assigned) it would be interesting to have seen how the authors address this possible bias.
Interestingly, the authors also find a slight inflation in fusion events over fission events across the tree (around 1 more per million years), although there are more fusion events nearer the root of the tree, with fission dominating nearer the leaves, and in particular, on the dense Drosophila subtree.
Finally, the authors performed a functional term enrichment analysis on the domain arrangements gained by fusion and fission events and showed that, in both cases, terms relating to signalling were significantly overrepresented in these populations, emphasising the potential importance that modular evolution may play in this area.
Detecting overlapping communities is essential to analyzing and exploring natural networks such as social networks, biological networks, and citation networks. However, most existing approaches do not scale to the size of networks that we regularly observe in the real world. In the paper by Gopalan et al. discussed in this week’s group meeting, a scalable approach to community detection is developed that discovers overlapping communities in massive real-world networks. The approach is based on a Bayesian model of networks that allows nodes to participate in multiple communities, and a corresponding algorithm that naturally interleaves subsampling from the network and updating an estimate of its communities.
The model assumes there are 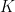
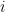
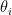
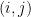
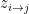
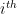
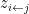

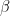

This model defines a joint probability 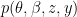

Community detection in a physics citation network.
One limitation of the method is that it does not incorporate automatic estimation of the number of communities, which is a general problem with clustering algorithms. Still, enabling sophisticated probabilistic analysis of structure in massive graphs is a significant step forward.
As it’s known in non-parametric kernel density estimation the effect of the bandwidth on the estimated density is large and it is usually the parameter who makes the tradeoff between bias and roughness of the estimation (Jones et.al 1996). An analogous problem for histograms is the choice of the bin length and in cases of equal bin lengths the problem can be seen as finding the number of bins to use. A data-base methodology for building equal bin-length histograms proposed by (Knuth 2013) based on the marginal of the joint posterior of the number of bins and heights of the bins. To build the histogram first the number of bins has to be selected as the the value (
where 

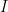
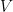


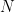
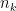
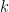
Now, the height (
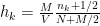
In the case of a normal distribution the authors suggest a sample of 150 data points to “accurately and consistently estimate the shape of the distribution”.
The following figure shows the relative log-posterior of the number of bins (left) and the estimated histogram for a mixture of three normal samples and a uniform [0,50] (right).

Knuth, K. H. (2013). Optimal data-based binning for histograms. arXiv preprint physics/0605197. The first version of this paper was published on 2006.
Jones, M. C., Marron, J. S., and Sheather, S. J. (1996). A brief survey of bandwidth selection for density estimation. Journal of the American Statistical Association,91(433), 401–407.

Intro
Antibodies have very well conserved structures and their binding site is chiefly comprised of the six CDRs. The great similarity between the 1700+ antibody structures that can be found in SAbDab/PDB prompted the introduction of numbering schemes which act as coordinates with respect to the sequence/structural features of antibodies. The earliest such numbering scheme was introduced by Wu and Kabat, followed by the structurally informed Chothia-scheme which was eventually amended by Abhinandan and Martin. Even though there are several of those schemes, the one currently endorsed by the World Health Organization (WHO) is this of IMGT.
The Program Downolad.
It annotates the framework and CDR residues according to three definitions: Kabat, Chothia or Contact. You can download it here.
Possible issues?
You need internet connection for this program to work since it calls the Abnum service, thus you should cite the following if you use this code:
Abhinandan, K.R. and Martin, A.C.R. (2008) Analysis and improvements to Kabat and structurally correct numbering of antibody variable domains Molecular Immunology, 45, 3832-3839.
How to use it?
As an example test case, type the following in the Framer directory:
python Framer.py --f 1A2Y.pdb --c AB --o my_first_output --d chothia
This should get the the heavy and light chains of 1A2Y (A and B) and leave the output in a folder called my_first_outupt.
Options:
–f: Antibody file
–c: Antibody chains (you can submit just one or several)
–o: Output folder name – NB this is going to be created in the directory you call Framer from!
–d: CDR definition to be used, possible options are: chothia, kabat and contact.
Output files:
There are four output files:
red_blue.pdb: The pdb with b-factor colored CDRs. The CDRs have B-factor of 100.00 and the framework 0.00.
paratope.txt: The CDR residues, given in the format [id][whitespace][chain]
framework.txt: The Framework residues, given in the format [id][whitespace][chain]
full_info.txt: Full breakdown of the annotation given in the format:
Original ID Original Chain AA Chothia ID CDR(FR=frame,or CDR id)
In last week’s journal club we delved into the history of Ramachandran plots (Half a century of Ramachandran plots; Carugo & Djinovic-Carugo, 2013).

Polypeptide backbone dihedral angles. Source: Wikimedia Commons, Bensaccount
50 years ago Gopalasamudram Narayana Ramachandran et al. predicted the theoretically possible conformations of a polypeptide backbone. The backbone confirmations can be described using three dihedral angles: ω, φ and ψ (shown to the right).
The first angle, ω, is restrained to either about 0° (cis) or about 180° (trans) due to the partial double bond character of the C-N bond. The φ and ψ angles are more interesting, and the Ramachandran plot of a protein is obtained by plotting φ/ψ angles of all residues in a scatter plot.
The original Ramachandran plot showed the allowed conformations of the model compound N-acetyl-L-alanine-methylamide using a hard-sphere atomic model to keep calculations simple. By using two different van der Waals radii for each element positions on the Ramachandran plot could be classified into either allowed regions, regions with moderate clashes and disallowed regions (see Figure 3 (a) in the paper).
The model compound does not take side chains into account, but it does assume that there is a side chain. The resulting Ramachandran plot therefore does not describe the possible φ/ψ angles for Glycine residues, where many more conformations are plausible. On the other end of the spectrum are Proline residues. These have a much more restricted range of possible φ/ψ angles. The φ/ψ distributions of GLY and PRO residues are therefore best described in their own Ramachandran plots (Figure 4 in the paper).
Over time the Ramachandran plot was improved in a number of ways. Instead of relying on theoretical calculations using a model compound, we can now rely on experimental observations by using high quality, hand picked data from the PDB. The way how the Ramachandran plot is calculated has also changed: It can now be seen as a two-dimensional, continuous probability distribution, and can be estimated using a full range of smoothing functions, kernel functions, Fourier series and other models.
The modern Ramachandran plot is much more resolved than the original plot. We now distinguish between a number of well-defined, different regions which correlate with secondary protein structure motifs.
Ramachandran plots are routinely used for structure validation. The inherent circular argument (A good structure does not violate the Ramachandran plot; The plot is obtained by looking at the dihedral angles of good structures) sounds more daring than it actually is. The plot has changed over time, so it is not as self-reinforcing as one might fear. The Ramachandran plot is also not the ultimate guideline. If a new structure is found that claims to violate the Ramachandran plot (which is based on a huge body of cumulative evidence), then this claim needs to be backed up by very good evidence. A low number of violations of the plot can usually be justified. The Ramachandran plot is a local measure. It therefore does not take into account that domains of a protein can exert a force on a few residues and just ‘crunch’ it into an unusual conformation.
The paper closes with a discussion of possible future applications and extensions, such as the distribution of a protein average φ/ψ and an appreciation of modern web-based software and databases that make use of or provide insightful analyses of Ramachandran plots.
This week’s topic of the Journalclub was about the prediction of potential T cell epitopes (=prediction of the binding affinity between peptides and MHC). In this context I presented the latest paper in the NetMHCpan series:
Karosiene E, Rasmussen M, Blicher T, Lund O, Buus S, Nielsen M. NetMHCIIpan-3.0, a common pan-specific MHC class II prediction method including all three human MHC class II isotypes, HLA-DR, HLA-DP and HLA-DQ. Immunogenetics. 2013 Oct;65(10):711-24
The reliable prediction of the peptide/MHC binding affinity is already a scientific aim for several years: Early approaches were based on the concept of anchor residues i.e. those peptide side-chain which are pointing in the direction of the MHC. The next “generation” of methods was matrix based i.e. it was simply counted how likely it is that a specific residue is at peptide position 1, 2, 3, … for binding and non-binding peptides of a specific MHC allele. In the next step methods started to incorporate higher order approaches i.e. positions within the peptide influence each other (e.g. SVM, ANN, …). While such methods are already quite reliable their major limitation is that they can only be trained on the basis of sufficient experimental binding data per allele. Therefore a prediction for alleles with only few experimental peptide binding affinities is not possible or rather poor in quality. This is especially import because there are several thousand HLA alleles: The latest version of the IPD – IMGT/HLA lists for example 1740 HLA Class I A and 2329 HLA Class I B proteins (http://www.ebi.ac.uk/ipd/imgt/hla/stats.html). Some of them are very frequent and others are not but this the aim would be a 100% coverage.
Pan specific approaches go one step further and allow the binding prediction between peptide and an arbitrary MHC allele for which the sequence is known. The NetMHCpan approach is one of them and is based on the extraction of a pseudo sequence (those MHC residues which are in close proximity to the peptide) per MHC allele. Subsequently this MHC pseudo sequence as well as the peptide sequences are encoded using some algorithm e.g. a BLOSUM matrix. Finally the encoded sequences and the experimental binding affinities are fed into an Artificial Neural Network (ANN). For an illustration of the workflow see:

http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2981877/bin/1745-7580-6-S2-S3-2.jpg
This approach turned out to be quite successful and (not too much) depending on the allele the area under ROC reaches in average a healthy 0.8x number which is already quite competitive with experimental approaches.
The netMHCpan series has progressed over the last years starting to cover MHC class I in humans (2007) and beyond (2009). Then MHC class II DR (2010) and in the latest version also DP and DQ (2013). With the next paper expected to be about non human MHC class II alleles the prediction of the binding affinity between peptides and pretty much every MHC should be possible.
