
OPIG recently acquired 55 additional computers all of the same make and model; they are of a decent specification (for 2015), each with quad-core i5 processor and 8GB of RAM, but what to do with them? Cluster computing time!

Along with a couple of support servers, this provides us with 228 computation cores, 440GB of RAM and >40TB of storage. Whilst this would be a tremendous specification for a single computer, parallel computing on a cluster is a significantly different beast.
This kind of architecture and parallelism really lends itself to certain classes of problems, especially those that have:
- Independent data
- Parameter sweeps
- Multiple runs with different random seeds
- Dirty great data sets
- Or can be snipped up and require low inter-processor communication
With a single processor and a single core, a computer looks like this:

These days, when multiple processor cores are integrated onto a single die, the cores are normally independent but share a last-level cache and both can access the same memory. This gives a layout similar to the following:

Add more cores or more processors to a single computer and you start to tessellate the above. Each pair of cores have access to their own shared cache, they have access to their own memory and they can access the memory attached to any other cores. However, accessing memory physically attached to other cores comes at the cost of increased latency.

Cluster computing on the other hand rarely exhibits this flat memory architecture, as no node can directly another node’s memory. Instead we use a Message Passing Interface (MPI) to pass messages between nodes. Though it takes a little time to wrap your head around working this way, effectively every processor simultaneously runs the exact same piece of code, the sole difference being the “Rank” of the execution core. A simple example of MPI is getting every code to greet us with the traditional “Hello World” and tell us its rank. A single execution with mpirun simultaneously executes the code on multiple cores:
$mpirun -n 4 ./helloworld_mpi
Hello, world, from processor 3 of 4
Hello, world, from processor 1 of 4
Hello, world, from processor 2 of 4
Hello, world, from processor 0 of 4
Note that the responses aren’t in order, some cores may have been busy (for example handling the operating system) so couldn’t run their code immediately. Another simple example of this would be a sort. We could for example tell every processor to take several million values, find the smallest value and pass a message to whichever core has “Rank 0” that number. The core at Rank 0 will then sort that much smaller number set of values. Below is the kind of speedup which was achieved by simply splitting the same problem over 4 physically independent computers of the cluster.

As not everyone in the group will have the time or inclination to MPI-ify their code, there is also HTCondor. HTCondor, is a workload management system for compute intensive jobs which allows jobs to be queued, scheduled, assigned priorities and distributed from a single head node to processing nodes, with the results copied back on demand. The server OPIG provides the job distribution system, whilst SkyOctopus provides shared storage on every computation node. Should the required package currently not be available on all of the computation nodes, SkyOctopus can reach down and remotely modify the software installations on all of the lesser computation nodes.




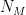 groups. Let
groups. Let 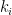 be the degree of node
be the degree of node 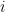 and
and  the number of links or edges to other nodes in the same group as node
the number of links or edges to other nodes in the same group as node  .
. ,
,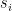 is the group node
is the group node 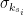 is the standard deviation of
is the standard deviation of 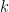 in such group.
in such group.






