If you ask someone who works with membrane protein structures what they think a helix kink is, you will probably get a fairly vague answer. They would probably tell you something like a kink is a point in the helix where there is a sharp change in the direction of the helix axis. Like the helix on the right in the picture.

Two example helices
They might also tell you that they frequently (or always) involve Proline, and that they feature the absence of some of the backbone hydrogen bonds that you would otherwise expect to observe in a helix. But that is about as far as the consensus goes.
There are a number of published methods to identify kinks in helices (ProKink, Helanal, MC-Helan, and TMKink, to name a few). The detail of identifying kinks (and hence the definition of kinks) differs a lot between the methods. That would be fine if they all identified the same helices as having kinks, and the same positions in each helix as kinked, but they do not. Even the number of helices that are kinked differs a lot – anything from 6 to 60%. It would be easy if all α-helices were like the two in the figure – clearly kinked or straight. Generally the methods agree on these ‘easy’ helices. But there are lots of helices that are difficult to classify.
Why do we want to computationally identify them? Well, kinks are functionally important in membrane proteins. Membrane proteins are both medicinally important, and hard to experimentally determine the structure of. So, modelling their structures is a useful thing to do, and their structure includes kinks. But also, they are a structural motif. If we can write down what we think a kink is, use that definition to identify kinks, and then use information from those kinks we identified to (reliably) predict the existence of other kinks, we will know that we fully understand to motif.
There are a remarkably large number of perfectly sensible ways to identify a sharp change in the axis of an α-helix, given the coordinates of the backbone atoms.
The published methods (mentioned above) are a bit cumbersome (they require you to create input files, and parse at least one, if not many, output files), so from my experience people tend to make their own methods (That is not just me with Kink Finder, but many of the people who need to identify kinks that I have spoken to at conferences do not use a published method).
A short summary of ways to identify kinks:
- Fit axes to short sections of helix (4-8 residues). Give each residue an angle calculated from the angle between the axis of the section before it, and the axis of the section after it. If the largest angle in the helix is greater than a threshold, annotate the helix as kinked, at the residue with the largest angle
- Methods similar to (1), but using a measure other than an angle. For example, the curvature of a line fitted to the helix, or the distance between the ith and (i+4)th Cα residue
- Identify sections of the helix that are ‘straight’. Where a helix contains more than on section, it is kinked
- Look at them, and classify
Even for (1), there are lots of ways to calculate a helix axis. You can us least-squares (very sensitive to the number of residues used in the fit), or fitting to a cylinder (better, but slower), or a vector method using 4 Cαs. Similarly for (2), you could fit a spline, or just a polynomial. This is before you decide on (e.g.) how many residues you are going to fit axes to (6?, 7? 8?), or what smoothing parameter to use in the spline fit, or what order polynomial.

How should we classify this helix?
The point is, there are lots of ways to do it, but there is no consensus definition. You can use one method, or 2, or more, but they will give you different answers. Because we have no concise definition, it is hard to say which helix classification is ‘right’ and which one is ‘wrong’. Take this helix, it is not perfectly straight, but is it not straight enough to be kinked? Or is the change in axis over a number of turns, making it curved rather than kinked?
There are a few ways round this problem, where your computer program is struggling to ‘correctly’ classify helices. Rather than using a computer, you could manually curate a set, which has been done. However, there are a few issues here. First, their definition of a kink was a bit vague – and certainly difficult to turn into a computer program. Second, humans are very good at seeing patterns, even when they are not really there. Third, the end aim, at least for me, is to incorporate this into structure prediction, where a computational formulation of a kink is necessary. Another solution is to accept that your program is deficient, and put the borderline (i.e. difficult) cases into a third group, and removing them from your analysis. This does help to improve your signal, but is a little unsatisfactory.
To conclude, there are not two types of helices, there is more of a spectrum, from very straight, through a bit kinked, to very kinked. Classifying them is a hard problem, with lots a different approaches available. But, while the methods give different answers (particularly in identifying the position of the kink in the helix), and they do not indicate the same causes of kinks, there is work to be done.

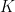
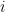
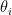
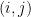
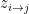
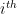
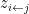

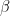

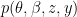
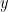
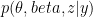



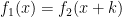 where k is the shift between the two distributions, thus if k=0 then the two populations are actually the same one. This test is based in the rank of the observations of the two samples, which means that it won’t take into account how big the differences between the values of the two samples are, e.g. if performing a WMW test comparing S1=(1,2) and S2=(100,300) it wouldn’t differ of comparing S1=(1,2) and S2=(4,5). Therefore when having a small sample size this is a great loss of information.
where k is the shift between the two distributions, thus if k=0 then the two populations are actually the same one. This test is based in the rank of the observations of the two samples, which means that it won’t take into account how big the differences between the values of the two samples are, e.g. if performing a WMW test comparing S1=(1,2) and S2=(100,300) it wouldn’t differ of comparing S1=(1,2) and S2=(4,5). Therefore when having a small sample size this is a great loss of information.






