
In cheminformatics, there are many possible ways to encode chemical data represented by small molecules and proteins, such as SMILES, fingerprints, chemical descriptors etc. Recently, utilising graph-based methods for machine learning have become more prominent. In this post, we will explore why representing molecules as graphs is a natural and suitable encoding.
What is a graph?
First we should discuss what we mean when we use the term graph. We are not referring to a plot of a function, but instead a graph, 
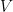
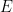
This representation can be made more complex by allowing vertices to have different types, different types of edges, edges that only point in one direction (i.e. 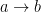

Representing molecules as graphs
Molecules are simply atoms joined together by bonds. These atoms may well be of different types, and the bonds might also be different, but this sounds a lot like a graph where the atoms are the vertices and the bonds are the edges of our graph!

Figure 1: Left – Toluene in standard chemical notation; Right – Toluene in a visual graph format.
Indeed, Figure 1 provides a simple visual example of the graph representation of a molecule, toluene (or methlybenzene). Since all atoms are carbon, it is possible to encode this molecule fully with a single atom type and two bond types (either single and aromatic, or single and double if kekulized). However, alternate atom typing is possible, for example taking separate atom types for aromatic carbons and aliphatic carbons. This highlights both the flexibility of a graph encoding but also the need to choose a representation, some of which may be more or less useful for the specific task.
What does a computer see?
Computationally, a graph is represented as two matrices: one for vertices, 

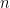
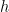

Using the example in Figure 1, taking two vertex types (aromatic & aliphatic carbon) and two edge types (single & aromatic), we can represent the graph by the matrices shown in Figure 2.

Figure 2: Matrix representation of toluene. Left – Vertex information; Right – Adjacency matrix.
Who cares?
We have described a simple, machine-readable format, that captures all basic features of a molecule, and is readily extendable to include any number of additional, user-defined vertex and edge features. Now that we have our molecular graph in matrix format, we can apply various graph-based machine learning methods on this. These are beyond the scope of this discussion, but could form the topic of a future blog post.