
At the group meeting on the 3rd of February I presented the results of the paper “A General Method for Insertion of Functional Proteins within Proteins via Combinatorial Selection of Permissive Junctions” by Peng et. al. This is interesting to our group, and especially to me, because this is a novel way of designing an antibody, although I suspect that the scope of their research is much more general, their use of antibodies being a proof of concept.
Their premise is that the structure of a protein is essentially secondary structures and tertiary structure interconnected through junctions. As such it should be possible to interconnect regions from different proteins through junctions, and these regions should take up their native secondary and tertiary structures, thus preserving their functionality. The question is what is a suitable junction? This is important because these junctions should be flexible enough to allow the proper folding of the different regions, but also not too flexible as to have a negative impact on stability. There has been previous work done on trying to design suitable junctions, however the workflow presented in this paper is based on trying a vast number of junctions and then identifying which of them work.
is important because these junctions should be flexible enough to allow the proper folding of the different regions, but also not too flexible as to have a negative impact on stability. There has been previous work done on trying to design suitable junctions, however the workflow presented in this paper is based on trying a vast number of junctions and then identifying which of them work.
As I said above their proof concept is antibodies. They used an antibody scaffold (the host), out of which they removed the H3 loop and then fused to it, using junctions, two different proteins: Leptin and FSH (the guests). To identify the correct junctions they generated a library of antibodies with random three residues sequences on either side of the inserted protein plus a generic linker (GGGGS) that can be repeated up to three times.
They say that the theoretical size of the library is 10^9 (however I would say it is 9*20^6), and the actually achieved diversity of their library was of size 2.88*10^7 for Leptin and 1.09*10^7. Next step is to identify which junctions have allowed the guest protein to fold properly. For this they devised an autocrine-based selection method using engineered cells that have beta lactamase receptors which have either Leptin or FSH as agonists. A fluoroprobe in the cell responds to the presence of beta lactamase producing a blue color, instead of green and therefore this allows the cells with the active antibody-guest designed protein (clone) to be identified using FRET-based fluorescence-activated cell sorting.
They managed to identify 6 clones that worked for Leptin and 3 that worked for FSH with the linkers being listed in the below table: 
There does not seem to be a pattern emerging from those linker sequences, although one of them repeats itself. For my research it would have been interesting if a pattern did emerge, and then that could be used as a generic linker for future designers. However, this is still another prime example of how  well an antibody scaffold can be used a starting point for protein engineering.
well an antibody scaffold can be used a starting point for protein engineering.
As a bonus they also tested in vivo how their designs work and they discovered that the antibody-leptin design (IgG-Leptin) has a longer lifetime. This is probably due to the fact that being a larger protein this is not filtered out by the kidneys.

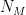 groups. Let
groups. Let 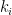 be the degree of node
be the degree of node 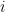 and
and  the number of links or edges to other nodes in the same group as node
the number of links or edges to other nodes in the same group as node  .
. ,
,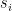 is the group node
is the group node 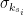 is the standard deviation of
is the standard deviation of 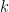 in such group.
in such group.















