
A simple toy example of a three layered network:
Note 1: In order to view the 3D plots, mac users will need Xquartz installed (https://www.xquartz.org/).
require(igraph)
require(rgl)
#Another package that might be needed is "rglwidget". The function writeWebGL will show an error stating if rglwidget is required.
######################################////
######The basics######################////
######################################////
#1) Create a "food" network (three layers)
set.seed(432)
g1<-watts.strogatz.game(dim = 1,size = 5,nei = 2,p = .5,loops = FALSE,multiple = FALSE)
g2<-watts.strogatz.game(dim = 1,size = 10,nei = 2,p = .2,loops = FALSE,multiple = FALSE)
g3<-watts.strogatz.game(dim = 1,size = 30,nei = 1,p = .5,loops = FALSE,multiple = FALSE)
g123=g1+g2+g3
#Create more edges btw layers
g123=rewire(g123,each_edge(prob=.4,loops = FALSE,multiple = FALSE))
ne=15;add_edges(g123,edges = cbind(sample(1:vcount(g1),size = ne,replace = TRUE), sample((vcount(g1)+1):vcount(g123),size = ne,replace = TRUE)))#top layer
ne=30;add_edges(g123,edges = cbind(sample((vcount(g1)+1):(vcount(g1)+vcount(g2)),size = ne,replace = TRUE), sample((vcount(g1)+vcount(g2)+1):vcount(g123),size = ne,replace = TRUE)))#second layer
#A quick plot of the graph
plot(g123,vertex.size=1,vertex.label.cex=0.02)
#Create 3d coordinates of the network layout
circpos=function(n,r=1){#Coordinates on a circle
rad=seq(0,2*pi,length.out=n+1)[-1];x=cos(rad)*r;y=sin(rad)*r
return(cbind(x,y))
}
#
lay=rbind(cbind(circpos(vcount(g1),r=1), runif(n = vcount(g1),-1,1)),
cbind(circpos(vcount(g2),r=2), runif(n = vcount(g2),6,7)),
cbind(circpos(vcount(g3),r=4), runif(n = vcount(g3),13,17))
)
#2d plot using the previous layout
plot(g123,vertex.size=5,vertex.label=NA,layout=lay[,c(1,3)])
plot(g123,vertex.size=1,vertex.label=NA,layout=lay[,c(1,2)])

#3D graph plot
#Add some colour to nodes and edges
nodecols=c(rep("red",vcount(g1)),
rep("blue",vcount(g2)),
rep("yellow",vcount(g3)))
edgecols=function(elist,cols,grouplist){
whatcol=rep(length(cols)+1,nrow(elist))
finalcol=whatcol
for(i in 1:nrow(elist)){
for(k in length(cols):1){
if( k * (length( intersect(elist[i,], grouplist[[k]]) ) > 0)){
whatcol[i]=min(whatcol[i], k )
}
}
finalcol[i]=cols[whatcol[i]]
}
return(finalcol)
}
#Open 3d viewer
rgl.open()
rglplot(g123, layout=lay,vertex.size=5,vertex.label=NA,vertex.color=nodecols,
edge.color=edgecols(elist=get.edgelist(g123,names = FALSE),cols=c("orange","green","pink"),grouplist=list(1:vcount(g1), (vcount(g1)+1):(vcount(g1)+vcount(g2)), (vcount(g1)+vcount(g2)+1):vcount(g123)) )
)
###Storing the plot in an html file###
dirfolder="..." #your dir
#rgl.open()#instead of rgl.open use open3d, in order to save the plot.
open3d()
rglplot(g123, layout=lay,vertex.size=5,vertex.label=NA,vertex.color=nodecols,
edge.color=edgecols(elist=get.edgelist(g123,names = FALSE),cols=c("orange","green","pink"),grouplist=list(1:vcount(g1), (vcount(g1)+1):(vcount(g1)+vcount(g2)), (vcount(g1)+vcount(g2)+1):vcount(g123)) )
)
#Fix the view
rgl.viewpoint(theta=90, phi=0)
#Save a static 2d image:
rgl.snapshot(paste(dirfolder,"a_png_pic_0.png",sep=""), fmt="png", top=TRUE)
#Save the plot in a .htlm file:
rglfolder=writeWebGL(dir = paste(dirfolder,"first_net3d",sep=""), width=700)
#The previous function should create a file called index.htlm inside the folder "first_net3d". By opening this file in a browser (with javascript enabled) the 3d plot will be displayed again.
#Also the following command will open the plot in the browser:
browseURL(rglfolder)
Note 2: In order to view the .htlm file javascript should be enabled in the browser. (Here is an example on how to do this for safari ).
Although not covered in the previous script, further options are available such as edge/vertex size and the ability to control independently each of the nodes and edges in the graph. Here is an example that makes more use of these options:

3d network representing a T cell receptor. Edges are coloured according to a relevant path found between the bottom green node and the upper red node cluster.

T cell receptor (in blue), binding to a peptide (in red).
 or equivalently with their dispersion
or equivalently with their dispersion  .
. , where
, where 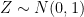 is an error term,
is an error term, 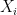 its the value recorded by instrument
its the value recorded by instrument 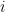 and where
and where 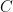 is the fixed true quantity of interest the instrument is trying to measure. But, what if
is the fixed true quantity of interest the instrument is trying to measure. But, what if  . Therefore the instruments would record quantities of the form
. Therefore the instruments would record quantities of the form 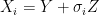 .
.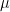 , the expected state of the phenomenon of interest is not a big challenge. Assume that there are
, the expected state of the phenomenon of interest is not a big challenge. Assume that there are  values observed from realisations of the variables
values observed from realisations of the variables 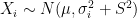 , which came from
, which came from 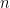 different instruments. Here
different instruments. Here 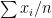 is still a good estimation of
is still a good estimation of 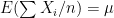 . Now, a more challenging problem is to infer what is the underlying variability of the phenomenon of interest
. Now, a more challenging problem is to infer what is the underlying variability of the phenomenon of interest  . Under our previous setup, the problem is reduced to estimating
. Under our previous setup, the problem is reduced to estimating  as we are assuming
as we are assuming 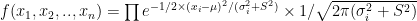 ,
,![\sum [(X_i- \mu)^2 - (\sigma_i^2 + S^2)] /(\sigma_i^2 + S^2)^2 = 0](https://s0.wp.com/latex.php?latex=%5Csum+%5B%28X_i-+%5Cmu%29%5E2+-+%28%5Csigma_i%5E2+%2B+S%5E2%29%5D+%2F%28%5Csigma_i%5E2+%2B+S%5E2%29%5E2+%3D+0&bg=ffffff&fg=000&s=0&c=20201002) .
.![E[\sum (X_i-\sum X_i/n)^2] = (1-1/n) \sum \sigma_i^2 + (n-1) S^2](https://s0.wp.com/latex.php?latex=E%5B%5Csum+%28X_i-%5Csum+X_i%2Fn%29%5E2%5D+%3D+%281-1%2Fn%29+%5Csum+%5Csigma_i%5E2+%2B+%28n-1%29+S%5E2&bg=ffffff&fg=000&s=0&c=20201002)
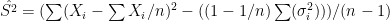 .
. and where the variance of the phenomenon of interest
and where the variance of the phenomenon of interest  . In the first scenario
. In the first scenario ![[10,1500^2]](https://s0.wp.com/latex.php?latex=%5B10%2C1500%5E2%5D&bg=ffffff&fg=000&s=0&c=20201002) , in the second from
, in the second from ![[10,1500^2 \times 3]](https://s0.wp.com/latex.php?latex=%5B10%2C1500%5E2+%5Ctimes+3%5D&bg=ffffff&fg=000&s=0&c=20201002) and in the third from
and in the third from ![[10,1500^2 \times 5]](https://s0.wp.com/latex.php?latex=%5B10%2C1500%5E2+%5Ctimes+5%5D&bg=ffffff&fg=000&s=0&c=20201002) . In each scenario the value of
. In each scenario the value of 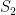 is estimated 1000 times taking each time another 200 realisations of
is estimated 1000 times taking each time another 200 realisations of  First simulation scenario where
First simulation scenario where  are shown by the blue (maximum likelihood) and red (alternative) histograms.
are shown by the blue (maximum likelihood) and red (alternative) histograms. First simulation scenario where
First simulation scenario where  First simulation scenario where
First simulation scenario where 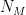 groups. Let
groups. Let 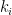 be the degree of node
be the degree of node  the number of links or edges to other nodes in the same group as node
the number of links or edges to other nodes in the same group as node  .
. ,
,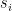 is the group node
is the group node 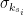 is the standard deviation of
is the standard deviation of 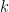 in such group.
in such group.


 These network comparison statistics create frequency vectors of subgraphs and then compare these frequencies between the networks to obtain an idea of the similarity of the networks relative to their subgraph counts.
These network comparison statistics create frequency vectors of subgraphs and then compare these frequencies between the networks to obtain an idea of the similarity of the networks relative to their subgraph counts.
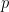 of being present).
of being present).
 . The probability of obtaining an edge between nodes i and j is given by
. The probability of obtaining an edge between nodes i and j is given by 

![[0,1]^d](https://s0.wp.com/latex.php?latex=%5B0%2C1%5D%5Ed&bg=ffffff&fg=000&s=0&c=20201002) . Now, given a radius or threshold distance (r), edges are drawn among nodes
. Now, given a radius or threshold distance (r), edges are drawn among nodes 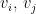
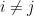 if
if  .
.

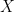 . In this article they proposed an exponential family of probability distributions to model
. In this article they proposed an exponential family of probability distributions to model 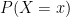 , where
, where  is a possible realisation of the random matrix
is a possible realisation of the random matrix  ) (see nodes 3-5 of the Figure below).
) (see nodes 3-5 of the Figure below).

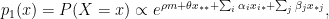
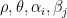 are parameters, and
are parameters, and  (identifying constrains).
(identifying constrains). 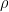 can be interpreted as the mean tendency of reciprocation,
can be interpreted as the mean tendency of reciprocation, 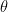 can be interpreted as the density (size) of the network,
can be interpreted as the density (size) of the network,  can be interpreted as as the productivity (out-degree) of a node,
can be interpreted as as the productivity (out-degree) of a node, 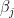 can be interpreted as the attractiveness (in-degree) of a node.
can be interpreted as the attractiveness (in-degree) of a node.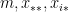 and
and 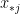 are: the number of reciprocated edges in the observed graph, the number of edges, the out-degree of node i and the in-degree of node j; respectively.
are: the number of reciprocated edges in the observed graph, the number of edges, the out-degree of node i and the in-degree of node j; respectively.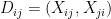 , the model assumes that all
, the model assumes that all  with
with 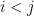 are independent.
are independent. and describe the joint distribution of
and describe the joint distribution of  , assuming all
, assuming all 
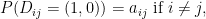
 .
.
 for
for 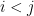 , and
, and 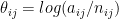 for
for  and
and  can be interpreted as the reciprocity and differential attractiveness, respectively. With a bit of algebra we get:
can be interpreted as the reciprocity and differential attractiveness, respectively. With a bit of algebra we get:![exp(\rho_{ij})=[ P(X_{ij}=1|X_{ji}=1)/P(X_{ij}=1|X_{ji}=0) ]/[ P(X_{ij}=1|X_{ji}=0) / P(X_{ij}=0|X_{ji}=0) ],](https://s0.wp.com/latex.php?latex=exp%28%5Crho_%7Bij%7D%29%3D%5B+P%28X_%7Bij%7D%3D1%7CX_%7Bji%7D%3D1%29%2FP%28X_%7Bij%7D%3D1%7CX_%7Bji%7D%3D0%29+%5D%2F%5B+P%28X_%7Bij%7D%3D1%7CX_%7Bji%7D%3D0%29+%2F+P%28X_%7Bij%7D%3D0%7CX_%7Bji%7D%3D0%29+%5D%2C+&bg=ffffff&fg=000&s=0&c=20201002)

 , and
, and  where
where 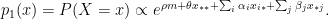
 ) that maximises the following posterior distribution for the number of bins:
) that maximises the following posterior distribution for the number of bins:
 is the number of bins,
is the number of bins,  is the data,
is the data, 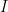 is prior knowledge about the problem, i.e. in particular the use of equal length bins and the range of data
is prior knowledge about the problem, i.e. in particular the use of equal length bins and the range of data 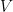 , which has the relation
, which has the relation  where
where  is the width of bins,
is the width of bins, 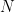 is the number of data points and
is the number of data points and 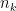 is the number of observations that fall in the
is the number of observations that fall in the  ) of the bins of the histogram is given by:
) of the bins of the histogram is given by: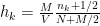 .
.
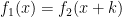 where k is the shift between the two distributions, thus if k=0 then the two populations are actually the same one. This test is based in the rank of the observations of the two samples, which means that it won’t take into account how big the differences between the values of the two samples are, e.g. if performing a WMW test comparing S1=(1,2) and S2=(100,300) it wouldn’t differ of comparing S1=(1,2) and S2=(4,5). Therefore when having a small sample size this is a great loss of information.
where k is the shift between the two distributions, thus if k=0 then the two populations are actually the same one. This test is based in the rank of the observations of the two samples, which means that it won’t take into account how big the differences between the values of the two samples are, e.g. if performing a WMW test comparing S1=(1,2) and S2=(100,300) it wouldn’t differ of comparing S1=(1,2) and S2=(4,5). Therefore when having a small sample size this is a great loss of information.
