
Protein-protein interaction (PPI) networks describe how proteins are connected to one another in terms of physical interactions. They can be used to aid our understanding of the individual roles of proteins (Sarajli ́c et al., 2013), the co-functioning properties of sets of proteins (West et al., 2013) and even the operation of the complete system (Janowski et al., 2014).
Different approaches have been proposed to analyse, describe and predict these PPI networks, such as network summary statistics, clustering methods, random graph models and machine learning methods. However, despite the large biological, computational and statistical interest in PPI net- works, current models insufficiently describe PPI networks (Winterbach et al., 2013; Ali et al., 2014; Rito et al., 2010). It is commonly accepted that proteins perform functions usually in conjunction with other proteins, forming a functional module (Lewis et al., 2010). Hence local structure is found to be important in protein-protein interaction networks (Planas-Iglesias et al., 2013).
Here we address the modelling problem locally by modelling the ego-networks of PPI networks by means of random graph models.
Random graph models
Loosely speaking, a random graph model is a set of rules that define an edge generation process among a set of nodes. Usually this set of rules relate to particular characteristics that are embedded in the network generation process. Here are three examples of such characteristics:
- Independence (each edge has a probability
of being present).
- Preferential attachment (nodes form edges with highly interacting nodes).
- Space-based interactions (an edge is present between two nodes if the distance between them small).
A classical model representing an independence structure is the ER(nv,p) model. This is a random graph on nv nodes, and where edges are present independently at random with probability p.

Now, the preferential attachment characteristic can be illustrated by the Chung-Lu model. That is, given an expected sequence of weights 


Finally, a model representing a spaced based network generation process could be the Geometric model. Here, nodes are placed uniformly at random in a d-dimensional square ![[0,1]^d](https://s0.wp.com/latex.php?latex=%5B0%2C1%5D%5Ed&bg=ffffff&fg=000&s=0&c=20201002)
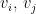
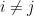


From the latter figures it can be seen that different models often lead to different network structures. Thus, although standard random graph models do not reproduce a sufficiently similar network structure to the one of PPI networks, they could still be good approximations for different local regions in a PPI network.
Finding a null model for PPI ego-networks
Our approach consist in finding local regions of the PPI networks that could be represented well by the random graph models. To do so, we propose to extract all 2-step ego-networks and classifying them according to some simple characteristic, network density for example.
Now, once the ego-networks of the PPI network have been extracted and binned according to their network density (ego-density). We assess the fit of the model to the PPI networks by comparing each bin of PPI ego-networks to the ego-networks extracted from a random graph model. This comparison is made by comparing the difference in the resulting number of subgraph counts, triangles for example, in each of the ego-networks within each bin.
The following figure illustrates the underlying idea of this procedure:

Following this approach we aim to find bins for which, possibly different models, reproduce similar subgraph counts as the ones obtained in the PPI ego-networks. However we expect to fin bins for which none of the standard models fit.
