
Do you use pandas for your data processing/wrangling? If you do, and your code involves any data-heavy steps such as data generation, exploding operations, featurization, etc, then it can quickly become inconvenient to test your code.
- Inconvenient compute times (>tens of minutes). Perhaps fine for a one-off, but over repeated test iterations your efficiency and focus will take a hit.
- Inconvenient memory usage. Perhaps your dataset is too large for memory, or loads in but then causes an OOM error during a mid-operation memory spike.

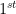 element to a
element to a 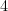 :
: