If you are a protein informatician, bioinformatician, biochemist, biologist or simply a person well informed about science, you probably heard about protein structure prediction. If that is the case, you might be wondering what all the fuss is about, right? If you never heard those terms before, don’t panic! You are about to find out what protein structure prediction is all about!
Based on my group meeting’s presentation last Wednesday, this blog entry will discuss why protein structure prediction is important and the potential limitations of existing methods. I will also discuss how the quality of input may be a potential source for lack of accuracy in existing software.
First, let us remember a little biology: our genetic code encrypts the inner-works of a complicated cellular machinery tightly regulated by other (macro)molecules such as proteins and RNAs. These two types of macromolecules are agents that perform the set of instructions codified by DNA. Basically, RNAs and proteins are involved in a series of processes that regulate cellular function and control how the genetic code is accessed and used.
For that reason, a huge chunk of genomic data can be pretty useless not that useful if considered on their own. Scientists around the globe have invested millions of moneys and a huge chunk of time in order to amass piles and piles of genome sequencing data. To be fair, this whole “gotta sequence ’em all” mania did not provide us with the fundamental answers everybody was hoping for. Cracking the genetic code was like watching an episode of Lost, in which we were left with more questions than answers. We got a very complicated map that we can’t really understand just yet.
For that reason, I feel obliged to justify myself: protein structures ARE useful. If we know a protein structure, we can formulate a very educated guess about that protein’s function. Combine that with empirical data (e.g. where and when the protein is expressed) and it can help us unveil a lot of info about the protein’s role in cellular processes. Basically, it can answer some of the questions about the (genomic) map. If only we could do that with Lost…
There is also evidence that knowing a protein’s structure can help us design specific drugs to target and inhibit that protein. Although the evidence of such biomedical application is sparse, I believe that with development of the field, there is a trend for protein structures to become more and more important in drug discovery protocols.
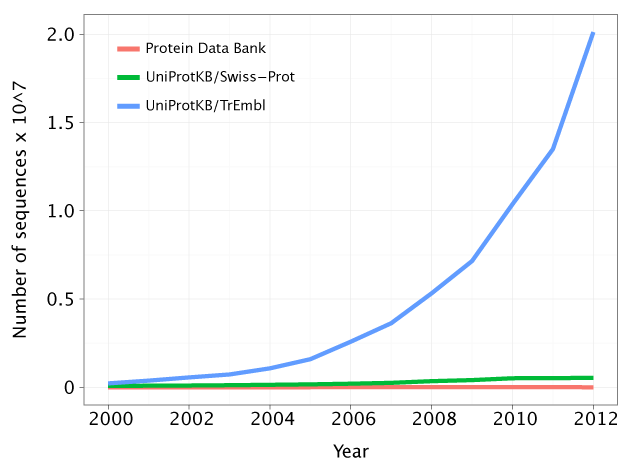
Still, if we look at the number of known genome sequences and known protein structures and at the growth of those figures over the past decade, we look at a drastic scenario:

There is a tendency for the gap between the number of protein sequences and protein structures to increase. Hence, we are getting more and more questions and little to no answers. Observe how the green line (the protein sequences associated with a known or predicted function) is very close to the red line (the number of known protein structures). However, there is a growing gap between the red and the blue line (the number of protein sequences). Source: http://gorbi.irb.hr/en/method/growth-of-sequence-databases/
Well, gathering protein structure data is just as important, if not more important, than gathering sequence data. This motivated the creation of Structural Genomics Consortiums (SGC), facilities that specialize in solving protein structures.
I am sorry to tell you that this is all old news. We have known this for years. Nonetheless, the graph above hasn’t changed. Why? The cost limitations and the experimental difficulties associated with protein structure determination are holding us back. Solving protein structures in the lab is hard and time consuming and we are far from being as efficient at structure determination as we are at genome sequencing.
There is a possible solution to the problem: you start with a protein sequence (a sequential aminoacid list) and you try to predict its structure. This is known as protein structure prediction or protein structure modelling. Well, we have a limited number of building blocks (20) and a good understanding of their physicochemical properties, it shouldn’t be that hard right?
Unfortunately, modelling protein structure is not as simple as calculating how fast a block slides on an inclined plane. Predicting protein structure from sequence is a very hard problem indeed! It has troubled a plethora of minds throughout the past decades, making people lose many nights of sleep (I can vouch for that).
We can attribute that to two major limitations:
1- There are so many possible ways one can combine 20 “blocks” in a sequence of hundreds of aminoacids. Each aminoacid can also assume a limited range of conformations. We are looking at a massive combinatorial problem. The conformational space (the space of valid conformations a protein with a given sequence can assume) is so large that if you could check a single conformation every nanosecond, it would still take longer than the age of the universe to probe all possible conformations.
2- Our physics (and our statistics) are inaccurate. We perform so many approximations in order to make the calculations feasible with current computers that we end up with very inaccurate models.
Ok! So now you should know what protein structure prediction is, why it is important and, more importantly, why it is such a hard problem to solve. I am going to finish off by giving you a brief overview of the two most commons approaches to perform protein structure prediction: template-based modelling (also known as homology modelling) and de novo structure prediction.
There is a general understanding that if two proteins have very similar sequences (namely, if they are homologs), than they will have similar structures. So, we can use known structures of homologs as templates to predict other structures. This is known as homology modelling.
One can do a lot of fancy talk to justify why this works. There is the evolutionary argument: “selective pressure acts on the phenotype level (which can encompass a protein structure) rather than the genotype level. Hence protein structures tend to be more conserved than sequence. For that reason and considering that sequence alone is enough to determine structure, similar sequences will have even more similar structures.”
One can also formulate some sort of physics argument: “a similar aminoacid composition will lead to a similar behaviour of the interacting forces that keep the protein structure packed together. Furthermore, the energy minimum where a certain protein structure sits is so stable that it would take quite a lot of changes in the sequence to disturb that minimum energy conformation drastically.”
Probably the best argument in favour of homology modelling is that it works somewhat well. Of course, the accuracy of the models has a strong dependency on the sequence similarity, but for proteins with more than 40% identity, we can use this method in order to obtain good results.
This raises another issue: what if we can’t find a homolog with known structure? How can we model our templateless protein sequence then? Well, turns out that if we group proteins together into families based on their sequence similarity, more than half of the families would not have a member with known structure. [This data was obtained by looking at the representativeness of the Pfam (a protein family database) on the PDB (a protein structure database).]
Ergo, for a majority of cases we have to perform predictions from scratch (known as free modelling or de novo modelling).
Well, not necessarily from scratch. There is a specific approach to free modelling where we can build our models using existing knowledge. We can use chunks of protein, contiguous fragments extracted from known structures, to generate models. This is known as a fragment-based approach to de novo protein structure prediction. And that is one big name!
One can think of this as a small scale homology modelling, where both the physics and evolutionary arguments should still hold true to some degree. And how do we do? Can we generate good models? We perform appallingly! Accuracies are too low to generate any useful knowledge in a majority of cases. The problem with the rare cases when you get it right is that you have no means to know if you actually got the right answer.
The poor quality of the results can be justified by the 2 biggest limitations discussed above. Yet something else might be in play. In homology modelling, if you use a bad template, you will most certainly get a bad model. In a similar way, using a bad set of fragments will lead you to a very poor final model.
Considering we already have the other two big issues (size of conformational space and accuracy of current potentials) to worry about, we should aim to use the best fragment library we possibly can. This has been the recent focus of my work. An attempt to make a small contribution to solve such a hard problem.
I would love to detail my work on finding better fragments here, but I believe this post is already far too long for anyone to actually endure it and read it until the end. So, congratulations if you made it through!















{kind=link}